
—— ผลการศึกษาความร่วมมือระหว่างศูนย์ควบคุมและป้องกันโรคเจ้อเจียง, Macro & Micro-Test และศูนย์ควบคุมและป้องกันโรคแห่งประเทศจีน ตีพิมพ์ในวารสาร Frontiers in Cellular and Infection Microbiology
ภาพรวมการศึกษา
ในเดือนพฤษภาคม 2026 วารสาร Frontiers in Cellular and Infection Microbiology (JCR Q1, IF ≈ 4.6) ได้ตีพิมพ์บทความวิจัยที่นำโดยศูนย์ควบคุมและป้องกันโรคประจำมณฑลเจ้อเจียง (Zhejiang CDC) โดยมีทีมชีวสารสนเทศจากบริษัท Beijing Macro & Micro-Test Bio-Tech Co., Ltd. และสถาบันควบคุมและป้องกันโรคติดต่อแห่งชาติ (China CDC) เป็นผู้ร่วมเขียน บทความวิจัยนี้มีชื่อเรื่องว่า:
“การระบุและวิเคราะห์ความสัมพันธ์ทางสายพันธุ์ของเชื้อ Brucella abortus จำนวน 7 สายพันธุ์ในมณฑลเจ้อเจียง ประเทศจีน”

การศึกษาครั้งนี้เป็นการวิเคราะห์การติดตามต้นกำเนิดทางวิวัฒนาการโดยใช้จีโนมทั้งหมดอย่างเป็นระบบครั้งแรกของเชื้อ Brucella abortus (B. abortus) ในมณฑลเจ้อเจียง ประเทศจีน ทีมวิจัยได้วิเคราะห์เชื้อแยก 7 ตัวอย่างที่เก็บรวบรวมตั้งแต่ปี 2015 ถึง 2025 (เชื้อที่มาจากมนุษย์ 4 ตัวอย่าง และเชื้อที่มาจากโค 3 ตัวอย่าง จากเมืองจินฮวา เมืองฉูโจว และเมืองหนิงโป) ผลการวิจัยให้หลักฐานทางจีโนมเกี่ยวกับต้นกำเนิดและเส้นทางการแพร่กระจายของ "สายพันธุ์ที่พบมากในภาคเหนือ" นี้ ในภูมิภาคที่มีการระบาดผิดปกติทางตอนใต้ของภาคตะวันออกของจีน
ภูมิหลังและความสำคัญ
โรคบรูเซลโลซิสเป็นโรคติดต่อจากสัตว์สู่คน เกิดจากแบคทีเรียในสกุล Brucella โดย Brucella abortus ส่วนใหญ่จะติดเชื้อในวัว แต่ก็สามารถก่อให้เกิดโรคในมนุษย์ได้เช่นกัน ในประเทศจีน โรคบรูเซลโลซิสมีความแตกต่างกันอย่างมากในแต่ละภูมิภาค โดยพบผู้ป่วยมากที่สุดในมณฑลทางภาคเหนือ (เช่น มองโกเลียใน ซานซี เฮยหลงเจียง) ในทางตรงกันข้าม มณฑลทางภาคใต้ รวมถึงเจ้อเจียง มีประวัติการระบาดของ Brucella melitensis เป็นหลัก โดยมีรายงานผู้ป่วย B. abortus น้อยมาก ความแตกต่างในระดับภูมิภาคนี้ทำให้การศึกษาลักษณะทางพันธุกรรมและการติดตามแหล่งที่มาของ B. abortus ในเจ้อเจียงเป็นเรื่องสำคัญลำดับต้นๆ ด้านสาธารณสุข
วิธีการและผลการค้นพบที่สำคัญ
ทีมวิจัยได้นำกลยุทธ์หลายด้านมาใช้ โดยผสมผสานชีววิทยาระดับโมเลกุลและชีวสารสนเทศเข้าด้วยกัน:
1.การระบุเชื้อก่อโรคและการจำแนกประเภทเบื้องต้น
การตรวจหาลำดับยีน BCSP-31 ด้วยวิธี PCR และ AMOS-PCR ยืนยันว่าเชื้อทั้งเจ็ดตัวอย่างเป็นเชื้อ B. abortus
การจำแนกสายพันธุ์โดยใช้ลำดับดีเอ็นเอหลายตำแหน่ง (MLST) โดยอิงจากยีนควบคุมการทำงานพื้นฐาน 9 ยีน พบว่าสายพันธุ์ทั้งหมดอยู่ในกลุ่มลำดับประเภท ST2 ซึ่งบ่งชี้ถึงความเหมือนกันทางพันธุกรรมสูงในกลุ่มสายพันธุ์ B. abortus ที่แพร่ระบาดในมณฑลเจ้อเจียง
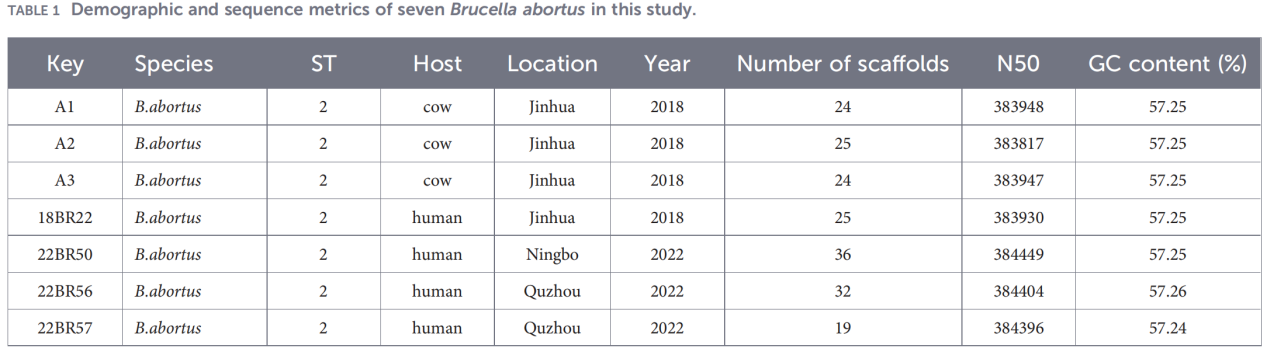
2.การวิเคราะห์ลักษณะทางพันธุกรรมทั้งจีโนม
การจัดลำดับจีโนมทั้งหมดดำเนินการบนแพลตฟอร์ม Illumina NovaSeq การวิเคราะห์ความเหมือนของนิวคลีโอไทด์เฉลี่ย (ANI) แสดงให้เห็นว่าสายพันธุ์ที่แยกได้จากมณฑลเจ้อเจียงมีความคล้ายคลึงกับสายพันธุ์อ้างอิง B. abortus 544 สูงถึง 99.99%
การวิเคราะห์แพนจีโนมเผยให้เห็นประชากรที่มีการอนุรักษ์สูง โดยระบุยีนหลักได้ 3,084 ยีน พร้อมกับยีนเปลือกเพียง 10 ยีน และไม่พบยีนแกนอ่อนหรือยีนเมฆเลย
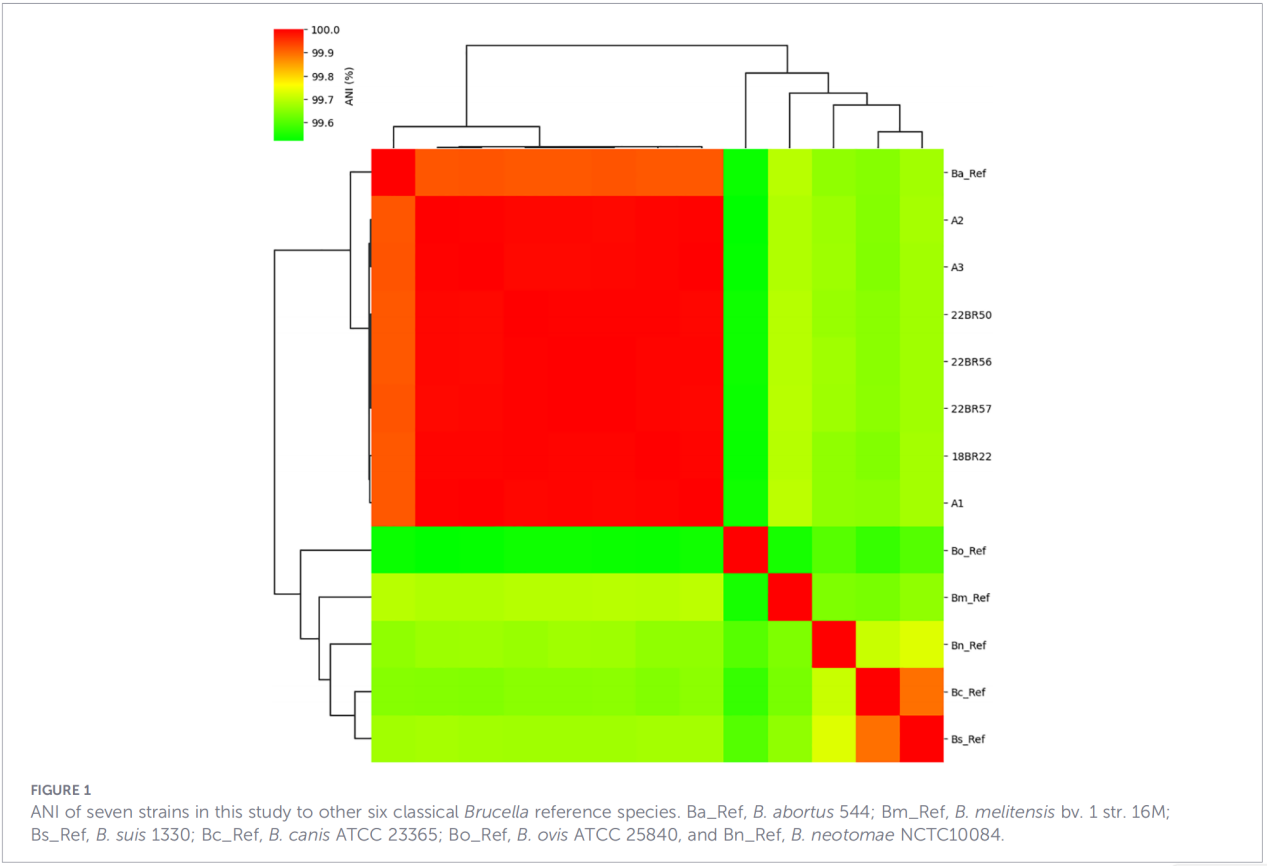
3.โปรไฟล์ยีนก่อโรคและยีนต้านทานยาปฏิชีวนะ
มีการคาดการณ์ปัจจัยที่เกี่ยวข้องกับความรุนแรงของโรคทั้งหมด 68 ปัจจัย ครอบคลุมเส้นทางคลาสสิก เช่น การสังเคราะห์ LPS ระบบการหลั่ง T4SS และระบบควบคุมแบบสององค์ประกอบ BvrR-BvrS ที่น่าสังเกตคือ ไอโซเลตทั้งหมดขาดจีนแอดฮีซิน bmaA และ btaF การวิเคราะห์ยีนต้านทานตรวจพบเฉพาะจีน mprF ในฐานข้อมูล CARD โดยไม่พบตัวกำหนดความต้านทานอื่น ๆ
 4. การสร้างแผนภูมิวิวัฒนาการและการติดตามการแพร่กระจาย
4. การสร้างแผนภูมิวิวัฒนาการและการติดตามการแพร่กระจาย
การวิเคราะห์โพลีมอร์ฟิสม์ของนิวคลีโอไทด์เดี่ยวในจีโนมหลัก (cgSNP) ทำให้ระบุตำแหน่งของสายพันธุ์จากเจ้อเจียงได้อย่างเฉพาะเจาะจงในแผนภูมิวิวัฒนาการระดับโลก ผลการวิจัยแสดงให้เห็นว่าสายพันธุ์จากเจ้อเจียงก่อตัวเป็นกลุ่มโมโนฟิเลติก (monophyletic group) ร่วมกับสายพันธุ์จากรัสเซีย มองโกเลีย และมณฑลทางตอนเหนือของจีนหลายแห่ง (หนิงเซี่ย เฮยหลงเจียง มองโกเลียใน เหอเป่ย กานซู ปักกิ่ง) กลุ่มนี้ยังแตกแขนงออกเป็นสามกลุ่มย่อยที่แตกต่างกัน (กลุ่มที่ 1–3) ซึ่งบ่งชี้ถึงเหตุการณ์การนำเข้าที่เกิดขึ้นอย่างอิสระหลายครั้ง
บทสรุปและนัยสำคัญ
งานวิจัยนี้เป็นการนำเสนอชุดข้อมูลจีโนมที่มีความแม่นยำสูงชุดแรกของ B. abortus ในมณฑลเจ้อเจียง และได้ข้อสรุปที่สำคัญหลายประการ:
- เคลar พื้นฐานทางพันธุกรรม– เชื้อ B. abortus ที่แพร่ระบาดในมณฑลเจ้อเจียงเป็นสายพันธุ์ ST2 มีโครงสร้างทางพันธุกรรมที่คงที่สูง และเป็นตัวแทนของสายพันธุ์โรคบรูเซลโลซิสในโคโดยทั่วไป
2. เอวีอัตราการแพร่ระบาดข้ามภูมิภาค– การวิเคราะห์ทางวิวัฒนาการไม่สนับสนุนการมีอยู่ของสายพันธุ์เฉพาะถิ่นที่เป็นอิสระในเจ้อเจียง แต่ข้อมูลกลับชี้ให้เห็นอย่างชัดเจนว่าสายพันธุ์เหล่านี้มีต้นกำเนิดมาจากทางตอนเหนือของจีน และอาจมีพื้นฐานทางวิวัฒนาการร่วมกันกับสายพันธุ์จากรัสเซียและมองโกเลีย การมีกลุ่มย่อยสามกลุ่มบ่งชี้ถึงเหตุการณ์การนำเข้าหลายครั้งแยกกัน
3. ผลกระทบต่อสุขภาพของประชาชน– ผลการศึกษาเน้นย้ำถึงคุณค่าของการเฝ้าระวังทางพันธุกรรมสำหรับโรคแท้งติดต่อ แม้ในภูมิภาคที่ไม่เคยมีการระบาดมาก่อน เช่น มณฑลเจ้อเจียง แม้จำนวนผู้ป่วยในปัจจุบันจะต่ำ แต่เครื่องมือที่มีความละเอียดสูง เช่น cgSNP สามารถติดตามแหล่งที่มาของการระบาดที่นำเข้าได้อย่างมีประสิทธิภาพ และให้หลักฐานทางวิทยาศาสตร์เพื่อหยุดยั้งห่วงโซ่การแพร่กระจายที่เกี่ยวข้องกับการขนส่งปศุสัตว์ข้ามมณฑล
งานวิจัยนี้ไม่เพียงแต่เติมเต็มช่องว่างทางการวิจัยในมณฑลเจ้อเจียงเท่านั้น แต่ยังให้ข้อมูลพื้นฐานใหม่สำหรับการเฝ้าระวังเชื้อโรคและการประเมินความเสี่ยงของโรคแท้งติดต่อในภูมิภาคสามเหลี่ยมปากแม่น้ำแยงซีอีกด้วย
ข้อมูลเกี่ยวกับเอกสาร:
Yang, Y., Shi, X., Chen, J., Wang, L., Wu, Z., Yao, W., … & Wu, B. (2026). การระบุและการวิเคราะห์เชิงวิวัฒนาการของเชื้อ Brucella abortus เจ็ดสายพันธุ์ในเจ้อเจียง ประเทศจีน Frontiers in Cellular and Infection Microbiology, 16, 1758965.
วันที่เผยแพร่: 10 มิถุนายน 2026